

Single cell analysis is transforming biology - but what lies beyond RNA? Single cell analysis has...
Single cell RNA sequencing (scRNA-seq) has revolutionized our ability to study cellular heterogeneity and gene expression at unprecedented resolution. However, one of the technical challenges that can compromise data quality and waste precious sequencing resources is the formation of multiplets. Understanding multiplets, how they arise, and their impact on experimental design is crucial for any researcher embarking on single cell studies.
What are multiplets?
In scRNA-seq experiments, a multiplet occurs when two or more cells share the same cell barcode. This results in a mixed transcriptional profile that combines gene expression from multiple distinct cells, representing a loss in the “single cell” nature of the data. Multiplets are the opposite of singlets, which represent the ideal scenario where exactly one cell is labeled per cell barcode.
Multiplets can range in complexity from doublets (two cells sharing a barcode) to triplets (three cells) and even higher-order multiplets such as quadruplets (four cells) or beyond. While doublets are the most common type of multiplet, higher-order multiplets become increasingly problematic as they create more complex mixed transcriptional profiles that are even more difficult to distinguish from genuine biological signals.
Multiplets pose a serious problem because they can potentially create misleading biological results. For instance, if two different immune cell types form a multiplet, the resulting expression profile might suggest the existence of a hybrid immune cell that expresses both cell type markers simultaneously (Zhang et al 2017). Without removing the compromised data, multiplets could lead to incorrect biological interpretations, spurious cell type classifications, and inflated estimates of cellular diversity.
How are multiplets detected?
The gold standard for determining multiplet rates of scRNA-seq methods is to mix cells from two different species (such as human and mouse) and then determine how often transcripts from both cell types are found assigned to the same cell barcode (Bloom 2018). This is sometimes called a “barnyard” experiment.
Here's how it works: Human and mouse cells are combined in equal proportions and processed to generate a scRNA-seq library. After sequencing, reads are mapped to both human and mouse reference genomes. Singlets will show reads mapping predominantly to one species (>95% typically), while multiplets will show significant contributions from both species. Due to the different permutations of potential cellular doublets – human:human, human:mouse, mouse:human, mouse:mouse - only two of which are observable, the actual multiplet rate is calculated as 2x the observed multiplet rate from a barnyard experiment. Calculating the final multiplet rate based on the observed rate is a critical, and sometimes missed, step when multiplet rates are reported.
While the “barnyard” approach provides a direct, quantitative measurement of the multiplet rate under specific loading conditions, most scRNA-seq samples are not a mixture of two species. To help deal with this challenge, several computational approaches have been developed to estimate multiplet rates from non-barnyard samples. Tools like DoubletFinder, Scrublet, and DoubletDetection work by simulating artificial doublets from the existing single cell data and training classifiers to distinguish these from real singlets (DePasquale et al 2019, McGinnis et al 2019, Wolock et al 2019). Additional tools include DoubletDecon, which uses deconvolution methods; Solo, which employs deep learning approaches; and cxds/bcds, which use co-expression and binary classification strategies respectively (Bais & Kostka 2020, Bernstein et al 2020). Typically, these methods identify potential problematic “cells” and remove them from the dataset.
The cost of multiplets: Wasted sequencing reads
Multiplets represent more than just a data quality issue – they also waste sequencing. Each multiplet consumes sequencing reads but provides unusable data that must be filtered out during analysis. This is particularly problematic given that sequencing typically represents a large fraction of the total cost of scRNA-seq experiments.
The waste is compounded because multiplets contain transcripts from multiple cells, leading to higher total UMI counts, and therefore much more sequencing waste than a “low quality” cell. A triplet, for example, might appear to have 3x more transcripts than a singlet, while a quadruplet could have 4x the transcript count. This means that higher-order multiplets not only waste more sequencing resources but also receive preferential allocation of sequencing depth, creating a compounding effect where the most problematic data consumes more valuable sequencing resources.
Genetic demultiplexing: A multiplet paradox
Genetic demultiplexing represents one approach for reducing per sample cost using droplet-based approaches. By pooling cells from multiple genetically distinct samples (different individuals, strains, or genetically barcoded cell lines) before single cell processing, researchers can dramatically reduce the number of scRNA-seq reactions required.
However, genetic demultiplexing creates a fundamental paradox with respect to multiplets. To maximize cost savings, researchers want to load as many cells as possible per reaction. But this high cell loading inevitably increases multiplet rates, leading to substantial data loss that partially negates the cost benefits.
In a typical genetic demultiplexing experiment, researchers might pool cells from 8-16 samples and load 50,000-100,000 cells to maximize throughput. At these loading densities, multiplet rates can reach up to 30%. While computational methods can identify and remove most multiplets by detecting cells with mixed genetic signatures, this represents an enormous waste of sequencing resources.
Optimizing experimental design
Understanding multiplet formation and its consequences enables more informed experimental design decisions.
For traditional droplet-based platforms, multiplet rates follow an approximately linear relationship with total cells analyzed. For every 1,000 cells, the multiplet rate increases by 0.4%. This means that if you recover 20,000 cells, approximately 8% of those cells are multiplets. When overloading the amount of cells per reaction, such as in the case of genetic demultiplexing, the multiplet rates can reach 30% at 100,000 cells. This linear scaling has profound implications for experimental design. While loading more cells increases throughput and reduces per sample and per cell costs, it comes at the expense of data quality through increased multiplet contamination.

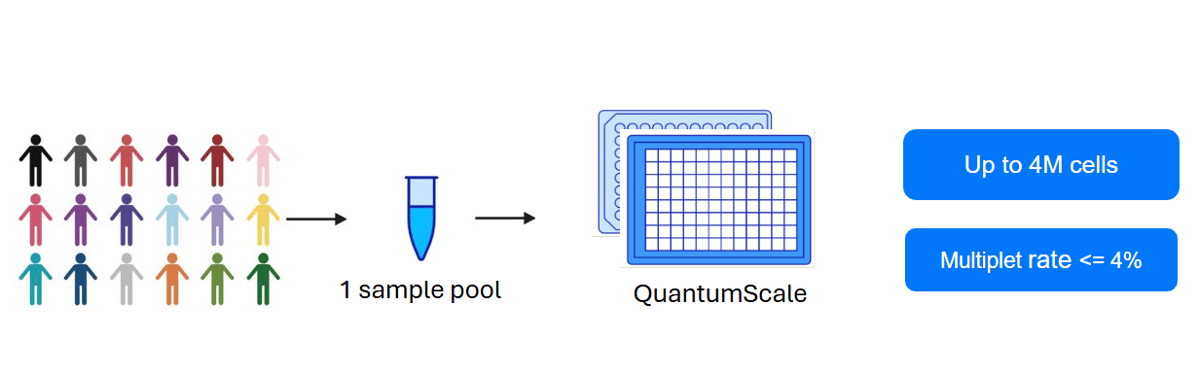
While droplet-based methods face an inherent trade-off between throughput and multiplet rates, massively parallelized barcoding approaches like QuantumScale RNA offer a different paradigm. These methods can achieve remarkably low multiplet rates even when processing millions of cells simultaneously. The key innovation lies in the combinatorial nature of cell labeling. Instead of relying on physical separation of individual cells, these methods use multiple rounds of barcoding where cells are distributed across different wells or compartments at each step. A cell's final barcode is the combination of all barcodes it receives across rounds. The probability that two cells receive identical barcode combinations across multiple rounds is extremely low, even with high cell densities. For example, with Quantum Barcoding technology, even when processing up to 4 million cells, the expected multiplet rate remains at or below 4%.

Conclusions
Multiplets represent a fundamental challenge in scRNA-seq that affects both data quality and experimental economics. The linear relationship between cell loading and multiplet formation creates an inherent trade-off that researchers must navigate when using droplet-based approaches. For experiments with large sample numbers and/or cell throughput, massively parallel barcoding methods such as QuantumScale RNA offer a promising solution which is not constrained by increasing multiplet rates.
As single cell technologies continue to evolve, addressing the multiplet problem will be crucial for realizing the full potential of population-scale studies and high-throughput applications. Understanding these principles enables researchers to make informed decisions about experimental design, budget allocation, and data interpretation in their single cell studies.
References
Bais AS, Kostka D. 2019. Scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics doi: 10.1093/bioinformatics/btz698
Bernstein NJ, Fong NL, Lam I, et al. 2020. Solo: Doublet identification in single cell RNA-seq via semi-supervised deep learning. Cell Syst doi: 10.1016/j.cels.2020.05.010
Bloom JD. 2018. Estimating the frequency of multiplets in single-cell RNA sequencing from cell-mixing experiments. PeerJ doi: 10.7717/peerj.5578
DePasquale EAK et al. 2019. DoubletDecon: Deconvolution doublets from single cell RNA-sequencing data. Cell Rep doi: 10.1016/j.celrep.2019.09.082
McGinnis CS et al. 2019. DoubletFinder: Doublet detection in single cell RNA sequencing data using artificial nearest neighbors. Cell Systems doi: 10.1016/j.cels.2019.03.003
Wolock SL et al. 2019. Scrublet: Computational idenfication of cell doublets in single cell transcriptomic data. Cell Systems doi: 10.1016/j.cels.2018.11.005
Zhang GXY et al. 2017. Massively parallel digital transcriptional profiling of single cells. Nat Comm doi: 10.1038/ncomms14049
Have questions or would like to talk to a single cell expert? Contact us below.

.png?height=200&name=Blog%20Featured%20Images%20(300%20x%20175%20px).png)
